
Safety & Quality
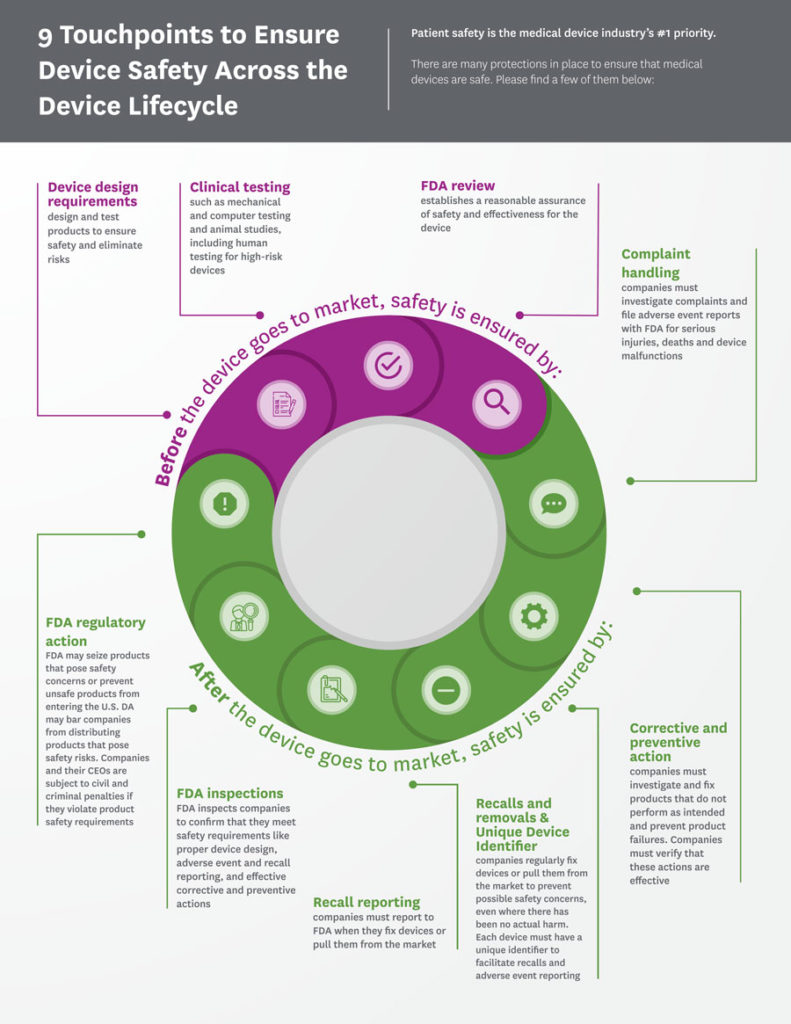
Patient safety is the medical device industry’s #1 priority. There are many protections in place to ensure devices are safe.
A Commitment to Safety, Effectiveness and Quality
The medical technology industry is committed to safety, effectiveness and quality for all devices. Companies employ scientists, physicians, engineers, quality experts and other professionals to research and examine every aspect of a medical device to ensure that it performs as intended.
Medical technology companies also meet stringent requirements set by FDA, whose oversight is considered the gold standard for regulation worldwide. FDA regulations cover the entire lifecycle of a medical technology – from design to manufacture to distribution – to assure that patients have access to safe, effective, and high-quality devices.
Ensuring Safety Throughout Development: Premarket Requirements
Safety, effectiveness and quality are the watchwords of the medical technology industry, and companies commit significant time and expertise to ensure that their devices perform as intended. This commitment starts at the earliest design phase and continues through the development, manufacturing and distribution of a device.
The universe of medical technology is vast – including everything from simple examination tools to sophisticated imaging and monitoring equipment – so FDA has developed a risk‑based premarket review system that determines the level of review based on the device type. Every year the agency subjects thousands of medical devices to extensive science-based reviews to ensure that they meet their pre-determined specifications. This approach is a model for medical technology regulation worldwide.
There are two main pathways to market:
- Low-to moderate-risk device types that have a well-established safety profile are cleared through the premarket notification or 510(k) process.
- Higher-risk technologies that often involve novel or first-of-a-kind innovations follow the premarket approval (PMA) application process.
Regardless of the pathway, FDA determines the evidence necessary before any new device or diagnostic can be deemed safe and effective, and the data that companies need to provide is comprehensive and thorough.
Continued Safety in the Field: Postmarket Requirements
Once a medical device undergoes FDA’s premarket review process, its manufacture and performance are tightly controlled and monitored through a series of postmarket requirements. FDA’s postmarket regulations encompass every aspect of a device’s manufacture – from production and processing to packaging and distribution – and require manufacturers to record, monitor and analyze their processes. As part of these requirements, medical device manufacturers must establish and maintain a comprehensive “quality system” to ensure their devices are manufactured consistently to specifications and have in place reporting mechanisms to detect, identify and mitigate device-related issues.
Mandatory adverse event reporting requirements provide a mechanism for manufacturers and other stakeholders to detect issues in a timely manner and report them to FDA. Medical device manufacturers must also establish comprehensive recall procedures so that if a device experiences a serious problem once on the market, it can be addressed quickly and effectively. Finally, for some higher-risk products, additional postmarket surveillance and tracking requirements may apply to provide an added level of safety monitoring.